Drugging KRAS: a Once ‘Undruggable’ Target
Edited by Tom Cohen
In 2020 there were over 19 million new cancer cases, accounting for about ten million deaths. 20% to 30% of all human tumours present mutations in the RAS subfamily of oncogenes. But according to Andreas Mantoulidis, Medicinal Chemistry Research Laboratory Head at Boehringer Ingelheim, they “have remained to be an elusive therapeutic target for over 40 years.”
- Using Innovative Approaches to Expand the Druggable Genome
- Target Identification, Phenotypic and Cell-based Screening
- Target Identification: Navigating the Intricate Environment of Human Biology
For example, K-RAS has been decidedly ‘undruggable’ after decades of searching due to the lack of bindable pockets in RAS proteins for small inhibitory molecules. However, as Mantoulidis explained, the possibility of drugging RAS proteins has had a renaissance. May 2021 saw FDA approval of Sotorasib, the first K-RAS inhibitor to do so, and since then others have achieved IND and clinical approval.
Boehringer Ingelheim’s Approach to Drugging K-RAS
Boehringer Ingelheim’s journey into tackling this target started ten years ago, collaborating with Vanderbilt University. All of their K-RAS hit finding was based on nuclear magnetic resonance (NMR) fragment screening, with programs including inhibitors for KRASG12C(off), KRASG12D(off), pan-KRAS (off), and pan-RAS (on).
Mantoulidis then answered why they would target mutant-selective K-RAS rather than casting a broader net with a pan-K-RAS inhibiting compound. “The reason is that we don’t know yet whether the pan-K-RAS approach will be tolerated in the clinic.” Furthermore, even when targeting only the G12C indication, this covers 13% of K-RAS cancers, which is still a significant figure.
Therefore, Mantoulidis’s team went with a fragment-based approach, targeting G12Ci in the switch II pocket. This is contrasting with the warhead-first approach employed by competitors; those approaches start with the warhead before digging deeper into the pocket to identify clinical candidates. The team at Boehringer Ingelheim instead try to find the best K-RAS binding motif with an MNR-based fragment screen, before developing and optimising the fragment further. Then finally, they find the optimal exit vector to address the G12C position with the covalent warhead function.
Fragment Based Approach Towards K-RAS Inhibition: BI-2852 in the Switch I/II Pocket
“KRAS is a switch, cycling between the on and the off state,” said Mantoulidis. “In a normal cell line, KRAS is off – resting and doing nothing.” He explained that as soon as a mutation occurs in KRAS, the tumour is in an active GTP-loaded form and signals downstream to induce cell growth and division. When KRAS is in its “on-state,” it presents a shallow lipophilic pocket called “switch I/II”. According to Mantoulidis, this pocket is very difficult to address with therapeutics to date: “it’s one of the hardest to target pockets that I’ve seen in my career so far.”
Mantoulidis explained that if you can address switch I/II with an inhibitor, the outcome should have acceptable selectivity and tolerability towards KRAS. After unsuccessful screening attempts of up to 1.7 million compounds, Boehringer Ingelheim switched to a fragment-first approach to address on-state KRAS.
NMR-based screening was employed and supplemented with X-ray confirmation of the binding site. The team did find some hits using this approach, but as Mantoulidis pointed out, the binding was very weak, and they were not able to measure the KD. What they were able to work out from the results was that each of the hits they found all had a common feature: an indole or indole-like fragment.
Hit Optimisation and SAR by Catalog
Mantoulidis’s team used this indole fragment as a starting point for their hit optimisation. “The next step was quite easy,” said Mantoulidis, “as we did not need to synthesise anything. We could just check the binding from the X-ray which we get quite early in the project.” Then they could ask: where in the fragment might they need to address in order to boost the potency further?
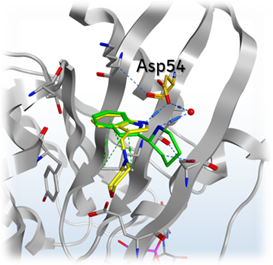
Mantoulidis pointed out the amino acid Asp54 on his slide and explained that the team thought they could address this part of the pocket with a salt in order to boost the potency into an acceptable range. This led them to use a SAR by catalog approach to test this hypothesis. This turned out to be successful as they ended up with new fragments with a measurable KD, although it was not potent enough to show up in biophysical assays and therefore the NMR approach was still required.
The team then continued to grow and develop their compound, improving its binding by establishing interactions with the KRAS pocket. They managed to get the molecule’s potency high enough to start measuring its affinity using biochemical assays.
They continued to grow the compound, optimising the binding, and trying to boost the potency. Mantoulidis’s team were unable to achieve a good enough selectivity between RAS isoforms which he described as “disappointing.” As a result, their investigations eventually led to a point where they thought they needed to stop the program or start from scratch.
Turning to Open Access Collaborative Research with opnMe
Because so much work had already been invested into the program, Boehringer Ingelheim decided to open up the molecule to research from third parties. They shared their compound, BI-2852 (along with its negative control compound, BI-2853), for free to the community on opnMe – a platform which offers compounds for free to the community in the interest of aiding research. opnMe shares well-characterised preclinical tool compounds openly so that researchers can order the compound and conduct experiments.
Making the molecule publicly available made sense to the company as KRAS is such a prevalent target, the more accessible research there is toward inhibiting it the better. The goal is to fulfil the potential of new small molecules through the power of collaboration. Hopefully, the full potential of these compounds will bring researchers one step closer to inhibiting cancers.
Join and network with over 200 industry leaders at Discovery US: In-Person, where we will address the latest advancements in target identification, validation and HIT optimisation.
Related Resources