







Process Development and Manufacturing Avenues for Gene Therapy Vectors

Presented by: Hemant Dhamne, Head of Process Development of Gene Therapy at KCL
Edited by: Cara Digby-Patel
The King's College Gene Therapy Vector Facility (GTVF) is a state-of-the-art facility for gene and cell therapy, with the capacity to manufacture a range of viral vectors, including lentiviral (LV), retroviral (RV), and adeno-associated vectors (AAV). Hemant Dhamne is the head of their process development team and is an expert in cell and gene therapy.
Speaking at Oxford Global's Gene Therapy Development &Manufacturing 2022: In-Person event, Dhamne talked us through the process of gene therapy vector production, specifically focusing on viral vector production.
Viral Vectors: An Overview
Genetic vectors are vehicles which allow for the targeted delivery of a therapeutic gene. Dhamne's work focuses on the process development and subsequent production of viral vectors, and his presentation offered insight into AAV and LV vector manufacture in particular.
- Combinatorial Culture Technology for Stem Cell Therapy Applications
- Upstream Bioprocessing
- Novel Technologies Advancing Downstream Bioprocessing
A viral vector system is created by stripping down the viral genome into its individual components and cloning into plasmids (see Figure 1). These plasmids, which contain the desired genes, or genes of interest (GOI), are transfected into a host system, such as HEK293. The host system, also referred to as the packaging cell line, is used to produce the virus which will deliver the GOI but will not replicate itself later.
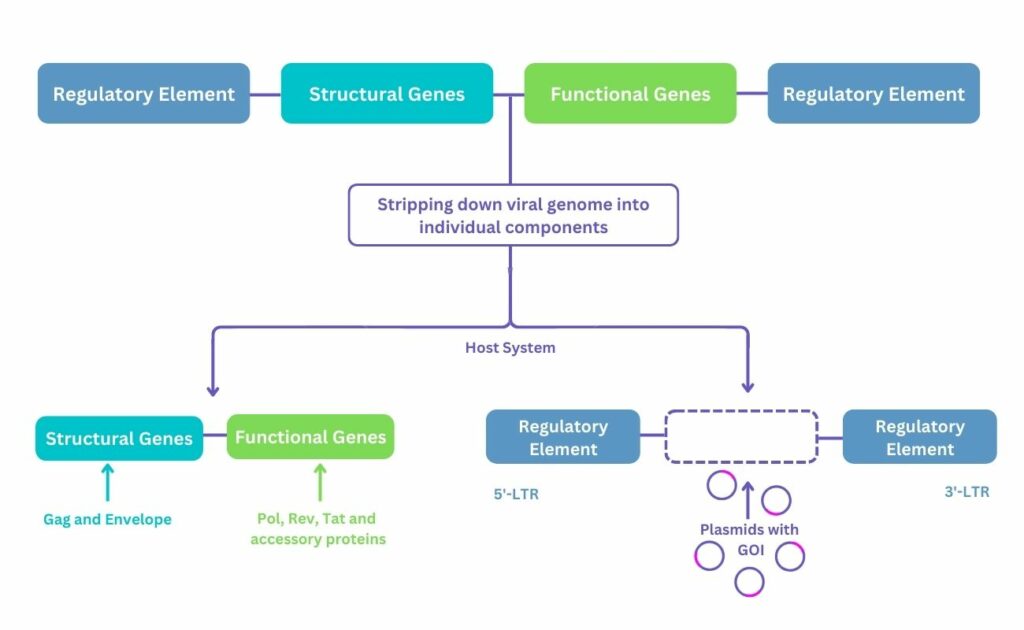
Utilising viral vectors as drugs presents several challenges. These arise partly due to their nature as complex entities; their molecule weight is in megadaltons, with AAV measuring 20 nm and whilst LV weighs 120 nm. Moreover, they are multimeric proteins containing nucleic acid at their core which are heterogeneous with respect to physical and functional titers. To add to this, enveloped viruses are also encased by a lipid covering with protein spikes.
Equally, the dosage requirement per patient is high. For a single batch of CAR-T manufacturing for one patient, around 1E+09 LV or RV particles are required. Similarly, for an in vivo gene therapy approach, 1E+13 to 1E+14 AAV particles are required per kilogram of patient. Therefore, the dose requirements are extremely high over several clinical trials, where there will be multiple approvals and multiple patients, which places more demand on an already challenging production process.
The Upstream Process: Packaging, Transfection, and Scalability
Firstly, a host cell line which will “package” the vector carrying the GOI into a delivery vehicle must be chosen. HEK293 cells are often used due to natural benefits, as they are easily transfected and support high-level expression of viral proteins. When selecting the host cell line, manufacturers must also consider whether they want adherent or suspension cells.
Transfection, the process of introducing the plasmid vectors, is the next stage. In viral vector production, transient transfection or co-transfection is usually performed. Viral vectors require multi-plasmid transfection. AAV vectors, for example, typically require three plasmids. Successful transfection of all plasmids must occur to produce functional vectors; if this does not happen, the result is low a titer and incomplete viral packaging.
A complexing reagent is also required for the transfection process, but these, too, present challenges. For example, calcium phosphate methods commonly have substantial batch-to-batch variability, whilst Liposome transfections are much more expensive, especially on the scale required for viral vector production.
Dhamne referenced the possibility of stable packaging cell lines, which would bypass the need for transient transfection. These cell lines could therefore eliminate certain challenges which arise from transfection; in particular, they could overcome issues which occur because such a low percentage of cells receive all the required plasmids via transfection. Though not yet readily available, stable packaging cell lines are growing field of research in the industry which holds a lot of promise.
The process must be scaled up following transfection to make it feasible in an industrial context. Different growth chambers are required for adherent and suspension processes. Dhamne noted that bed bioreactors are becoming increasingly popular for adherent processes because they are more scalable and convenient than cell factories in terms of ease of handling. Stirred-tank bioreactors and wave bioreactors are well-established methods for suspension processes in the biopharma and vaccine industries. They are now increasingly used in the viral vector industry too, as they are easily scalable in a linear fashion.
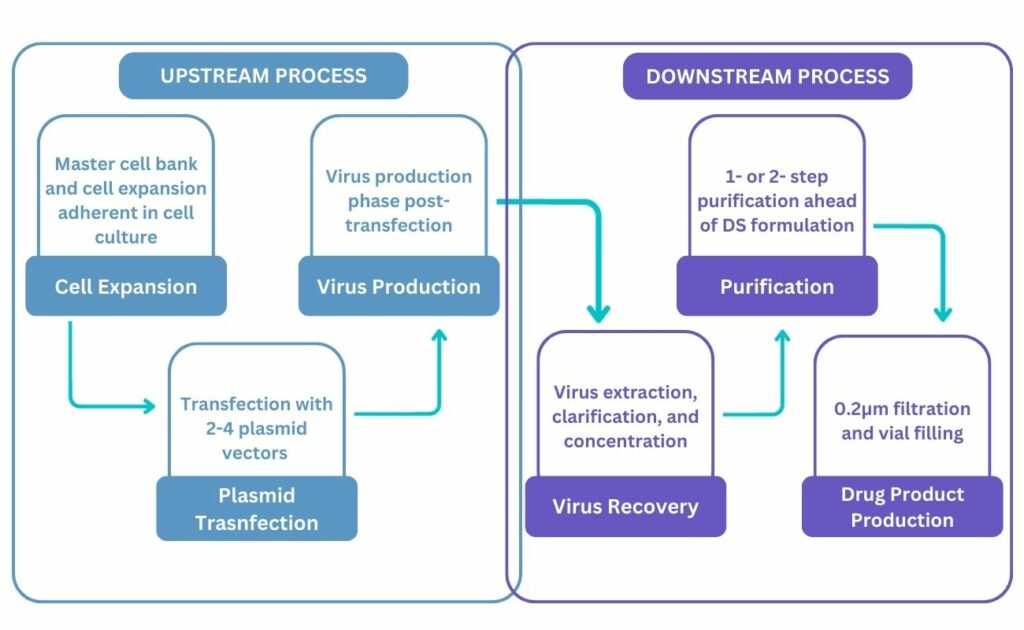
The Downstream Process: Virus Recovery and Purification
Once the host cells have produced high quantities of the virus, it is necessary to harvest and purify the viral particles. Centrifugation or crossflow filtration provide a sufficient means of harvesting cells. However, as AAV vector production is both intracellular and secretory for certain serotypes, difficulties may arise as large proportions of the virus remain within the cell.
These difficulties can be overcome using lysis, which is essentially the breakdown of a cell by disrupting its plasma membrane. Cell lysis typically uses mild detergents, mechanical stress, and freeze-thaw procedures to release the virus. Either during lysis or post-purification, the product is treated with a nuclease such as Benzonase in order to remove host cell DNA and RNA, residual plasmids and free-floating vector DNA/RNA, to minimise the nucleic acid contaminants.
The purification process can be performed either in one or multiple stages depending on the viral vector being manufactured. Centrifugation and microfiltration are typically used to remove cell fragments and debris for AAV vectors. Dhamne mentioned that, unlike Benzonase, a proteinase cannot be added to remove host cell proteins, so affinity chromatography is the best method to capture vector particles, as it allows the remaining impurities to be removed in flowthrough. However, affinity chromatography only results in partial purification: whilst it can generate high yields of purified AAV, it cannot differentiate between empty and full viral capsids, and the former impacts the safety and efficacy of AAV vectors.
Therefore, a second purification step may be required, either ion exchange chromatography or ultracentrifugation, to separate the full and empty capsids. Though this second purification is crucial to the manufacture of effective AAV vectors, it is not an easy process, as the scalability can pose issues and the equipment is expensive.
Future Outlook
Dhamne concluded with a discussion of how the manufacturing process could be rendered more effective, stating that “process efficiency, associated cost drivers and regulatory requirements all need to be addressed before we can establish the effective industrialisation of cell and gene therapies.”
Ultimately, refining the process lies in four key areas: single-use technology, faster analytics, automation and integration, and digitalisation. Dhamne closed by saying that if these aspects could be incorporated into viral vector production, most of the substantial issues within the field could be addressed, paving the way towards commercialisation and mass production.
Want to find out more about the latest stem cell therapy news? Register now for Oxford Global‘s 3D Cell Culture congress incorporates key trends and innovative technologies to accelerate the adoption of 3D models in preclinical research via advanced development, validation and application strategies.
Related Resources

