Using Innovative Approaches to Expand the Druggable Genome
At Discovery Europe 2022, Ulrich Hommel, Head of Integrated Lead Discovery at Novartis, stated that “Ever since the first human genome sequencing, scientists have asked how big is the druggable genome? And are we just drugging the tip of the iceberg?” His presentation at the Berlin conference set out to investigate this question and outlined the methods that Novartis have been taking to expand what is considered the druggable genome.
To consider the question as to whether current therapeutic targets are indeed the ‘tip of the iceberg,’ Hommel divided the genome into four classes of target (Oprea et al Nature Rev. Drug Discovery 17, 317 (2018)):
- Tclin: Targets with low molecular weight (LMW) drugs and understanding of their mode of action
- Tchem: targets with LMW inhibitors but no understanding of their mode of action
- Tbio: targets without LMW drugs but good link to disease
- Tdark: targets not belonging to Tclin, Tchem, or Tbio
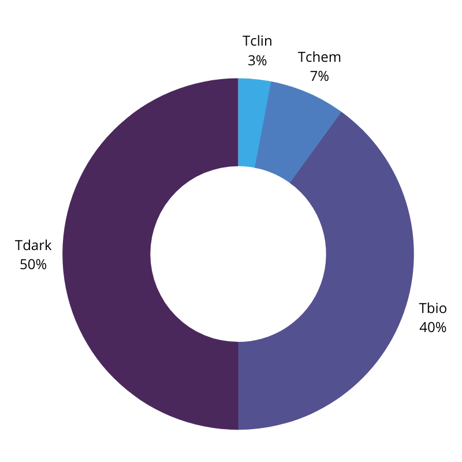
According to this study, Tclin targets make up only 3% of the druggable genome: Tchem targets make up 7%, while Tbio targets make up nearly 40%. This leaves 50% of the human genome as potential therapeutic targets. Therefore, there are plenty of targets in the genome that could potentially be drugged – the question is how to identify them.
One example of a Tbio target that Hommel gave was leukocyte function-associated antigen 1 (LFA-1) – a heterodimeric cell surface receptor. LFA-1 engages with its ligand ICAM-1 when it has been activated at its headpiece, also known as ?L-I domain. This is because LFA’s ?-chain contains a metal-ion dependent adhesion site (MIDAS), required for receptor binding.
Druggability Assessment of LFA-1 – Fragment Based Approach
Hommel explained that a phenotypic screen of LFA-1 yielded a molecule that could inhibit the interaction between ICAM and LFA-1. Interestingly, when looking at the models using NMR and X-ray, they saw that the compound had unexpectedly bound to a crevice that they had not anticipated to have any biological function. “The biological function of the protein was always considered to occur close to the magnesium site,” explained Hommel.
The molecule in question that had bound to this crevice was the cholesterol lowering agent lovastatin. Then, Hommel stated the thesis of the presentation by asking “do we really need to use phenotypic screens?” He exemplified this question by pointing out that he had done an unrelated fragment screen of the same protein with “28 or so compounds”. Hommel showed that the fragment screen had indeed returned a weak hit, noting that the chemical shift differences for the lovastatin and the hit fragment were very similar.
- Up-and-Coming Investigations in Drug Discovery and Development: Neuroscience and AI
- Target Identification, Phenotypic and Cell-based Screening
- Target Identification: Navigating the Intricate Environment of Human Biology
“In a nutshell, the phenotypic screening hit, and the fragment had the same binding site,” said Hommel. This means that fragment-based screening could reveal binding sites with biological function.
The In Silico Approach
Hommel further elaborated on how the compound affects LFA-1. LFA-1 includes an ?-helix – ?7 – which exists in a metastable state. This means ?7 is stable in its current conformation until a large enough change is introduced to its environment. When the receptor’s ligand binds to its I-domain, the ?7 helix moves downward, activating the receptor. Therefore, when inhibited, the helix can no longer move, and the receptor does not activate.
Hommel moved forward in the timeline of research in this area: “A group from Cambridge, UK used molecular dynamic simulations to see whether they could see conformations that resemble the states that we have seen in the X-ray structure.” Hommel showed the findings of this investigation (Figure 2), “they found three different populations: a conformation that resembles a low affinity state (LA), one with an intermediate affinity (IA), and, in a large population, the allosterically inhibited form (AI).”
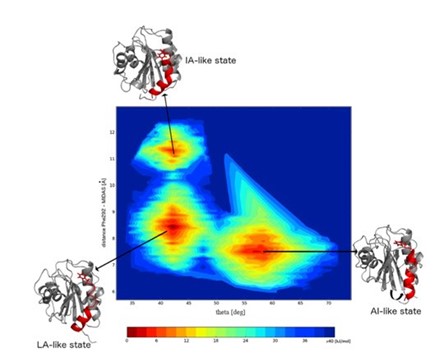
Hommel confirmed that by using computational analysis and artificial intelligence, it was possible to predict the states of proteins that can be inhibited.
Fragment Screening for a “Modern Target” – Interleukin-1 beta
“Can we learn from this and apply our findings to a modern target?” asked Hommel, introducing interleukin-1 beta (IL-1?), a pro-inflammatory cytokine. Hommel described how CANTOS, a study taking place in 2017, demonstrated that a monoclonal antibody targeting IL-1? has the potential to treat a number of diseases. For example, the cytokine has been linked to rheumatoid arthritis, gout, and periodic fevers, among others. “So, a low molecular weight inhibitor for this cytokine would be very interesting to have.”
“like a horseshoe grabs a ball.”
According to Hommel, there were two factors holding back IL-1?’s potential as a LMW therapeutic target. First, “from a crystallographic perspective, the cytokine seems undruggable.” Second, Hommel explained how IL-1?’s mode of action requires it binding to receptors using two binding sites, “like a horseshoe grabs a ball.” Hommel asked whether a LMW compound would ever be able to interfere with such a binding situation.
Using a Larger Screening Library
Hommel’s team used fragment-based screening to do this, utilising a massive library of ~4,000 fluorinated compounds named the LEF4000 library. The fragments are assembled in 132 mixtures containing up to 32 compounds each. Hommel pointed out that for IL-1?’s fragment screen, one compound showed a reduction of the signal intensity – “which is a sign for the reduced flexibility and mobility of this fragment.” This reduction is caused by the interaction with the protein, meaning that the team had found a hit.
The next step that Hommel’s team took was to analyse the hit in more detail. The NMR data showed that interestingly, the fragment had a slower binding event than what was typical for a fragment. Investigating further, the Novartis team mapped the binding site of the protein and discovered that the binding site is closed by two loops – “so perhaps these loops move during the binding event?” The protein structure solved via X ray crystallography confirmed that this was the case, and one of the loops was displaced.
Investigating the Binding Event
Was this just a silent or an artificial binding event? Hommel said that his intuition begged to differ: “During my 30-year career in drug discovery, I have learned that whenever you have slow conformational changes going on, then something important is going on in nature.” And indeed, this turned out to be correct, the binding site was very close to the interaction site of the receptor.
But why was this the case? Hommel outlined two possible explanations. First, induced fit – when the compound binds to the protein, the loop closes and covers the binding site. Second, conformational selection – this occurs when two conformations of the protein exist side-by-side and the compound prefers just one of them. However, when the team went looking for the alternate conformations, they were unable to find them.
Making the Invisible Visible and Exploiting Excited States
Hommel said that he and his team then turned to the work of Lewis Kay, who had studied a technique which would be able to reveal ‘invisible’ excited states in a protein. Hommel exemplified this with c-ABL, a kinase that had already been shown to exist in two excited states as well as its ground state. Importantly, each of these states harboured binding sites for a different inhibitor, so “all of these excited states are relevant, they are not just an artifact of nature. And if we find them, we can exploit them.”
“Can we prove that IL-1? has these excited states?” Hommel asked. Indeed, when employing the same technique as Lewis Kay, they were able to show that IL-1? existed in an excited state when subject to higher temperatures. With this information at their disposal, the team then mapped the excited state and were finally able to show the protein harbouring the binding site for the compound that they had seen before. Hommel rounded off the talk by concluding that, even though it was a lot of work, investigating ‘invisible’ excited states was worthwhile for drug discovery. This is because, not only does it help increase understanding of the therapeutic targets for disease, but it also increases understanding of the druggable genome.
Join 200+ leaders, experts and scientists helping to shape the future of drug discovery and development at Discovery US: In-Person.
Related Resources