Advances in Long Read DNA Sequencing

The road to long read DNA sequencing has been long and difficult. Over 30 years ago, in October 1990, the human genome project started. Its goal was to sequence and assemble the entire human genome. This was a time when sequencing was performed with radioactive sulfur or long acrylamide gels, and next-generation sequencing was associated with fluorescent key pillory sequences. Since then, our understanding of the human genome has been completely transformed. We can see that most people are genetically similar, and 99% of our genomes are exactly the same. Unfortunately, the most critical regions for disease often lie within that divergent one per cent.
What Is Long Read Sequencing?
The genome of humans is too long to be sequenced as one continuous string. Instead, next-generation short-read sequencing breaks up DNA into fragments that are then amplified and sequenced to produce reads. Bioinformatic techniques are then used to piece together these reads into an interpretable genomic sequence. Long Read Sequencing allows much longer reads and is split into two main types.
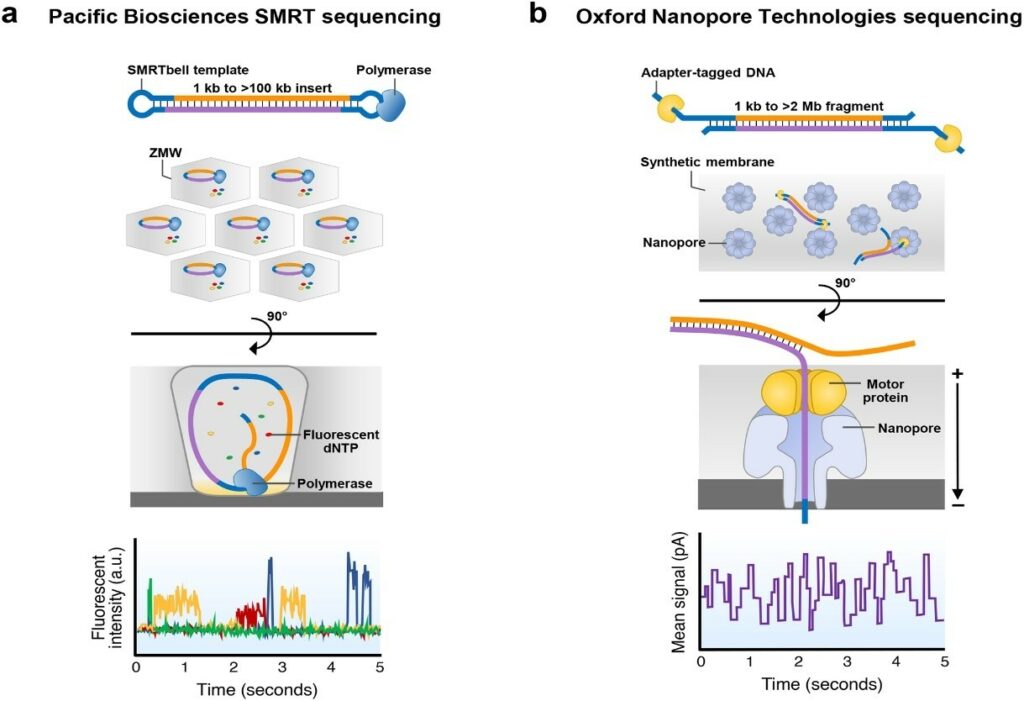
Synthetic long-read sequencing approaches use short-read sequencing and specialized computational tools to reconstruct longer reads. In addition, third-generation sequencers often avoid the need for amplification and can process DNA in real-time. This process, also known as “true” long-read sequencing, presents advantages over traditional short reads.
Currently, there are two primary producers of “true” long-read sequencing technologies: Pacific Biosciences (PacBio) and Oxford Nanopore Technologies (Nanopore).
Overcoming Challenges: Understanding Spacing and Distribution of Genes
One of the most critical questions in genomics is how many complex mendelian diseases are caused by the spacing and distribution of genes rather than non-coding variants. This is a tricky question to answer using only a simple reference. A read that spans the breakpoint may not align to the correct region. The flanking sequences will not be long enough to include the transgene as part of the reference; it must be added as an extra chromosome to the assembly.
Tools that can identify chromosomal translocations can give you the integration event in a VCF (Variant Call Format). However, if you have multiple integration events, understanding the phasing of the structure and the changes at each locus can be extremely difficult. Such problems become even worse if tandem or inverted integration events occur at the same location.
Alignment data in such cases, even with long reads, can be challenging to interpret unambiguously, and local failure genome assembly may become necessary. Also, if you do not have effective integration, the coverage that can be attained by whole genome assembly quickly gets out of reach, as does the amount of genomic DNA needed to have a reasonable likelihood of observing and interpreting frequency.
Advances in Targeted Long Read DNA Sequencing
The easiest method of finding a rare sequence in the genome is PCR. If you are confident of a sequence that lies at both the start and end of your region of interest, then you can design primers that amplify sequences of up to five kb. Such amplicons can be extended up to around ten kb. These kinds of approaches also allow easy multiplexing. One example of this is the addition of a column specific barcode at the five prime ends of the forward primers and a row specific barcode to the reverse primers. In this way, 384 well plates can be uniquely indexed by ordering only 40 oligos.
However, 384 amplicons are not an efficient use of a modern instrument. So usually, one would perform plate-based multiplexing using PacBio SMRT Bell indexing strategy where a single library is made from each plate. This will allow you to sequence up to 12,288 amplicons. In practice, this scale is more extensive than most people require. Nevertheless, it does allow amplicon sequencings projects to be performed with relative ease.
Issues With Long Amplitude Sequencing (PCR)
Unfortunately, the two primary requirements of PCR pose a significant problem for an important subset of experiments. There is a risk of only amplifying the molecules that are working as intended while missing where it failed due to poor parameters. As a result, many researchers wish they could do PCR with only one prime.
Accuracy and Error Rates: Room for Improvement
While the accuracy of both PacBio and Nanopore sequencing platforms has increased with incredible speed, accuracy is still far from perfect. There remains a lot of potential to improve the accuracy of long read data through improvements and informatics. There are also a number of improvements in informatics coming down the line for examples Google's work on DeepVariant.
DeepVariant is an analysis pipeline that uses a deep neural network to call genetic variants from next-generation DNA sequencing data. The algorithm development on the CCS calling is showing improvements in some of the repetitive regions in PacBio and Oxford Nanopore sequencing
Final Thoughts and Conclusion
Long Read DNA Sequencing has the potential to help diagnose several diseases and disorders. Recent advancements mean that long-read sequencing is no longer an expensive, low throughput, error-prone process. Today, it can allow an extremely high throughput processing of samples to be performed cheaply and efficiently in parallel. In addition, the read quality now far exceeds what can be obtained by shorter read technologies, making it an excellent choice for the phasing, and calling of genomic biomarkers. The next step would be a fully phased graph-based reference, and such a thing is becoming closer to reality. For more on the latest sequencing technologies, consider joining us for our upcoming NextGen Omics UK: In Person event.